Background
Two sample Mendelian randomisation is a method to estimate the causal effect of an exposure on an outcome using only summary statistics from genome wide association studies (GWAS). Though conceptually straightforward, there are a number of steps that are required to perform the analysis properly, and they can be cumbersome. The TwoSampleMR package aims to make this easy by combining three important components
- data management and harmonisation
- the statistical routines to estimate the causal effects
- connection to a large repository of the actual GWAS summary statistics needed to perform the analyses.
The general principles (Davey Smith and Ebrahim 2003; Davey Smith and Hemani 2014), and statistical methods (Pierce and Burgess 2013; Bowden et al. 2015) can be found elsewhere, here we will just outline how to use the R package.
This package uses the ieugwasr package to connect to the IEU OpenGWAS database of thousands of complete GWAS summary data.
Installation
If you are using macOS or Windows you can install a binary version of TwoSampleMR from our https://mrcieu.r-universe.dev with the following code.
install.packages("TwoSampleMR", repos = c("https://mrcieu.r-universe.dev", "https://cloud.r-project.org"))If you are using the latest long term support version of Ubuntu Linux (currently 24.04 Noble Numbat) you can install a binary version of TwoSampleMR with the following code.
options(HTTPUserAgent = sprintf(
"R/%s R (%s)",
getRversion(),
paste(getRversion(),
R.version["platform"],
R.version["arch"],
R.version["os"])
))
install.packages(
'TwoSampleMR',
repos = c(
'https://mrcieu.r-universe.dev/bin/linux/noble/4.5/',
'https://p3m.dev/cran/__linux__/noble/latest',
'https://cloud.r-project.org'
)
)To install TwoSampleMR from source from our GitHub repository run the following code.
# install.packages("remotes") # Run if remotes package not installed
library(remotes)
install_github("MRCIEU/TwoSampleMR")TwoSampleMR can be installed in WebR using the following code.
install.packages('TwoSampleMR',
repos = c('https://mrcieu.r-universe.dev', 'https://repo.r-wasm.org'))Overview
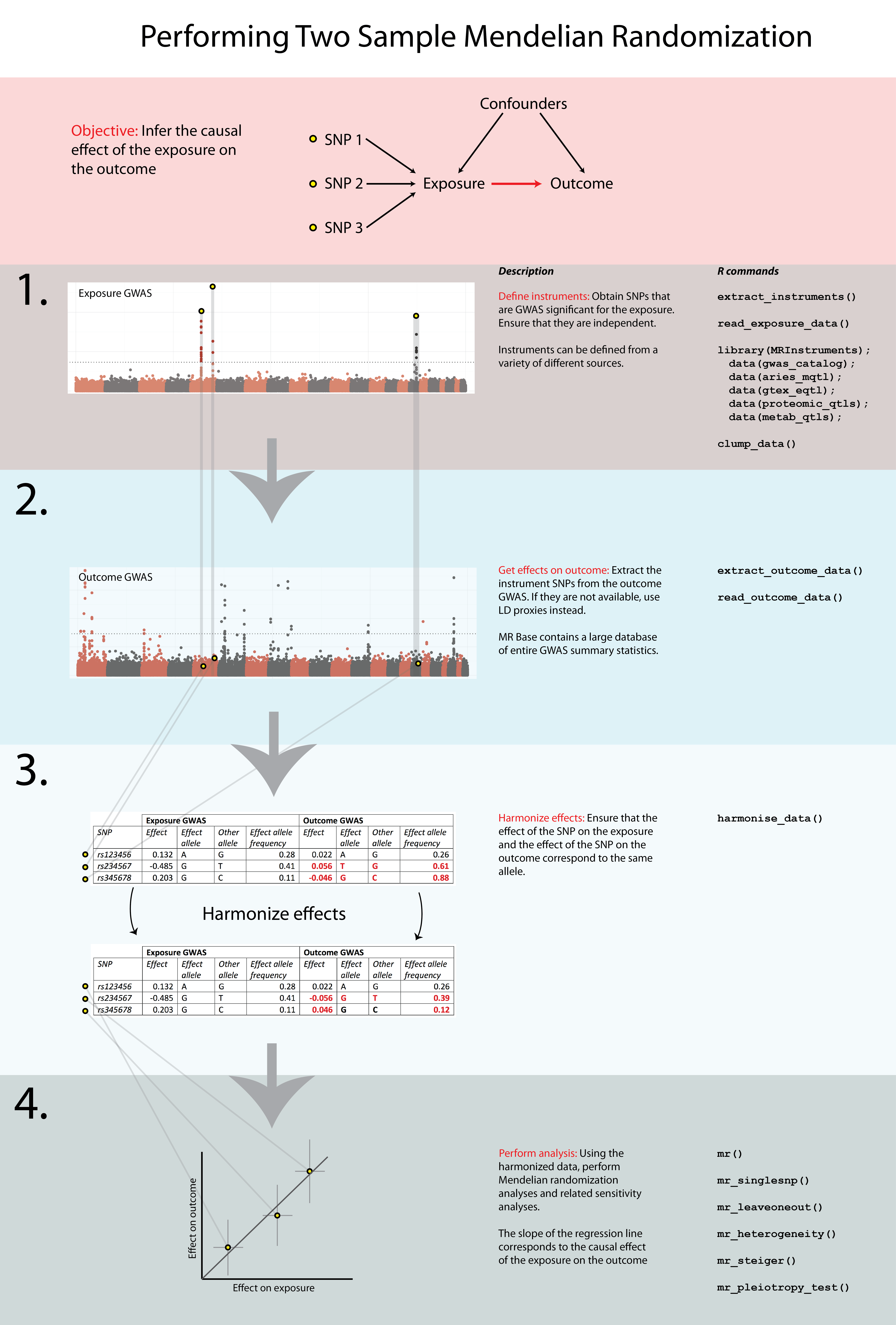
The workflow for performing MR is as follows:
- Select instruments for the exposure (perform LD clumping if necessary)
- Extract the instruments from the IEU GWAS database for the outcomes of interest
- Harmonise the effect sizes for the instruments on the exposures and the outcomes to be aligned to the same reference allele
- Perform MR analysis, sensitivity analyses, create plots, compile reports
A diagrammatic overview is shown here:
A basic analysis, e.g. the causal effect of body mass index on coronary heart disease, looks like this:
library(TwoSampleMR)
# List available GWASs
ao <- available_outcomes()
# Get instruments
exposure_dat <- extract_instruments("ieu-a-2")
# Get effects of instruments on outcome
outcome_dat <- extract_outcome_data(snps = exposure_dat$SNP, outcomes = "ieu-a-7")
# Harmonise the exposure and outcome data
dat <- harmonise_data(exposure_dat, outcome_dat)
# Perform MR
res <- mr(dat)Each step is documented on other pages in the documentation.
Authentication
The statistical methods in TwoSampleMR can be used on any data, but there are a number of functions that connect to the OpenGWAS database for data extraction. These OpenGWAS data access functions require authentication.
Authentication is changing The main differences are that:
- Authentication is required for most queries to OpenGWAS for everyone (i.e. no more anonymous usage)
- We are no longer using Google Oauth2. This has been replaced by a simple API key system.
Detailed information is given here: https://mrcieu.github.io/ieugwasr/articles/guide.html#authentication.